Hp6890 Gc-5973 Ms;
Hp-5MS毛细管色谱柱(30m*0.25mm*0.25μm);
载气:99.999%氦气;
环已烷、乙酸乙酯、丙酮均为国产分析纯试剂,用前重蒸;
2.4mol/L盐酸溶液(取20ml分析纯盐酸用重蒸水稀释至100ml);
萃取剂:环已烷和乙酸乙酯(4+1);
衍生化试剂:乙酸酐和吡啶(1+1);
0.2mol/L碳酸钾溶液(称取27.6g碳酸钾溶于重蒸水,稀释至1000ml);
五氯酚标准(购于中国标准技术开发公司标样开发部)。
1.2 实验条件
进样口温度:280℃;检测器温度:280℃;
柱温程序:初温170℃,保留2min,然后以10℃/min的升温速率,升至240℃,保持3min;
传输管温度;280℃;
EI电子源,电子能量为70ev;
载气流速;1.0ml/min;
选择离子监测方式,监测离子Sim(M/Z):266,308,237,165,130;
定量离子:266。进样量:1.0μl
1.3 标准溶液配制
标准贮备溶液:称取五氯酚标准0.0100g,用丙酮溶解,定容至100.00ml,此溶液的浓度为100μg/ml;
标准使用溶液:吸取标准贮备溶液1.0ml于100ml容量瓶中,用重蒸水稀释至刻度,此溶液的浓度为1.0μg/ml;
1.4 样品处理方法
1.4.1水样的采集和保存:水样采集后应尽快分析,如不能立即分析,应于每升水样中加入1ml硫酸,5g硫酸铜,置于冰箱中保存。
1.4.2 水样的预处理:取50ml水样于50ml比色管中,用2.4mol/L的盐酸溶液调PH<2(约0.3ml),加入萃取剂2.0ml,振摇1min,静置分层后,取出1.0ml有机相于5ml比色管中,加入少许无水硫酸钠,再加入50ul衍生化试剂,于60℃水浴中放置15min,冷却后,加入0.5ml碳酸钾溶液,充分混匀后,吸出有机相,待测。
1.4.3 标准曲线的制作:取50ml比色管7只,分别加入O,10,25,50,100,250,500微升标准使用溶液,加重蒸水至50ml,也就是含五氯酚0,0.20,0.50,1.0,2.0,5.0,10.0ug/L的标准系列,以下按(1.4.2)处理。
1.5.4 测定 先测定标准系列并绘制标准曲线,后进样品测定。
2.结果与讨论
2.1 定性和定量
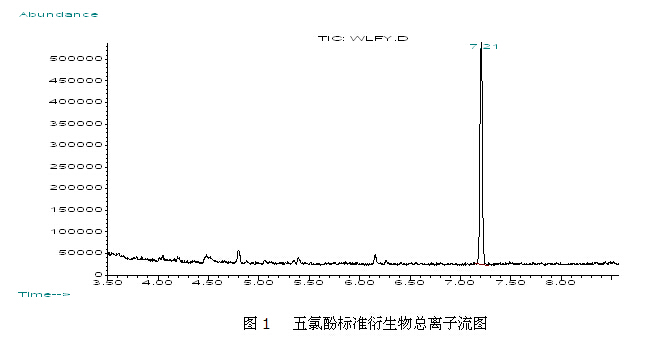
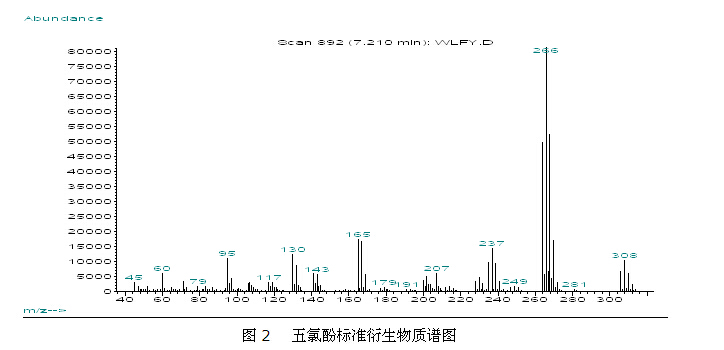
在实验条件下,五氯酚衍生物的保留时间为7.21分钟。用保留时间和特征离子266,308,237,165,130相结合定性,这比气相色谱单靠保留时间定性要准确,定量离子选择丰度最大的266,用外标法、峰高定量,五氯酚标准衍生物总离子流图见图1,质谱图见图2。
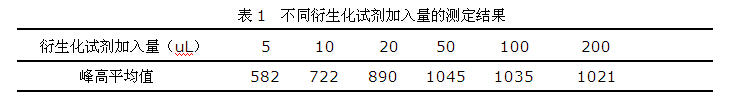
2.2 衍生化试剂加入量的选择 取50ml比色管6只,各加入50ml水样,100ul标准使用溶液,以下按(1.4.2)操作,衍生的时候,分别加入衍生化试剂5,10,20,50,100 ,200 ul,测定结果见表1。当加入50ul衍生化试剂时,标准峰高最大,故选择50 ul为衍化生化试剂加入量。
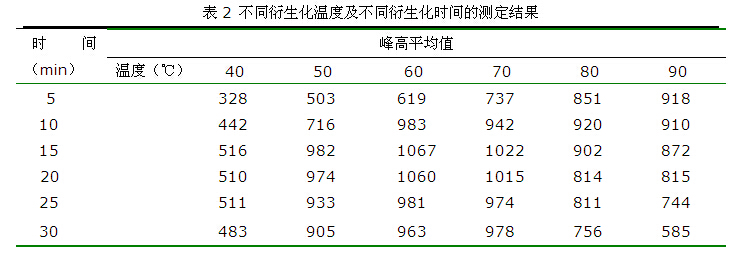
2.3 最佳衍生化温度及衍生化时间的筛选 不同衍生化温度与不同衍生化时间对衍生率产生不同影响。本文对含五氯酚2.0ug/L的合成水样在40、50、60、70、80、90℃不同衍生化温度及5、10、15、20、25、30 min不同衍生化时间条件下进行了实验测定,结果见表2。结果表明以60℃,15min为最佳分析条件。
2.4 碳酸钾溶液的洗涤次数 在电子捕获-毛细色谱法中,由于ECD检测器易污染,碳酸钾溶液至少清洗2次。而用气相色谱/质谱法测定,清洗一次就够了,较为简单。
2.5 标准曲线和检出限:按(1.4.3)操作,以峰高定量,每个浓度标准进样三次取平均值结果为O,105,267,536,1067,2631,5282。经回归计算,相关系数r=1.0,回归方程为:y=10.549x+4.066;以三倍噪声计,检测下限为0.0005ng,若取50ml水样分析,检测下限浓度为0.02μg/L。
2.6 精密度试验 分别配制含0.2,2.0,10 ug/L五氯酚标准的样品液,每个浓度各测定6次,计算相对标准偏差,结果见表3。
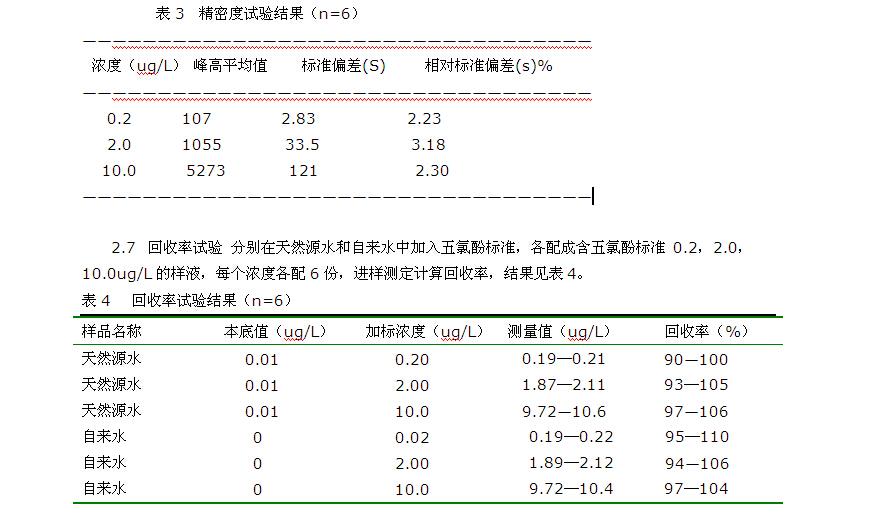
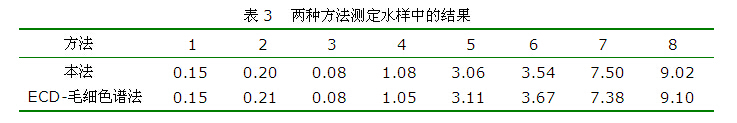
2.8 方法比对试验 取8份合成水样,同时用本法与电子捕获-毛细色谱法测定,结果经统计学处理(配对t检验,P> 0.05),两种方法测定结果差异无显著性。
内容仅供参考

 订购电话
订购电话 邮箱合作电话投诉与反馈售后服务及咨询
邮箱合作电话投诉与反馈售后服务及咨询 地址
地址 京公网备案 11010502037132号
京公网备案 11010502037132号
